
02209 Anhang IX MDR: Konformitätsbewertung auf der Grundlage eines Qualitätsmanagementsystems und einer Bewertung der technischen Dokumentation
|
Anhang IX MDR beschreibt im Detail die Aktivitäten zur Konformitätsbewertung basierend auf einem vollständigen Qualitätsmanagementsystem und einer Bewertung der technischen Dokumentation für die zu bewertenden Medizinprodukte. Viele der hier beschriebenen Tätigkeiten sind ebenfalls Bestandteil der Konformitätsbewertungsverfahren nach den Anhängen X und XI.
Bei der Erläuterung der in Anhang IX getroffenen Festlegungen und Anforderungen werden nicht nur die bis dahin bekannten Guidance-Dokumente, u. a. der MDCG, sondern auch die Festlegungen aus Anhang VII MDR (Von den Benannten Stellen zu erfüllende Anforderungen) berücksichtigt.
Die Kommentierung erfolgt abschnittsweise, um die Übersichtlichkeit und die Rückführbarkeit der Erläuterungen auf die regulatorische (Grund-)Anforderung besser darzustellen. Quelle des Textes ist die konsolidierte Fassung der MDR vom 24.04.2020.
Verweise im Werk auf die EN ISO 13485:2016 sind unverändert auch für die harmonisierte Fassung der Norm in der Ausgabe von 2021 gültig. Arbeitshilfen: von: |
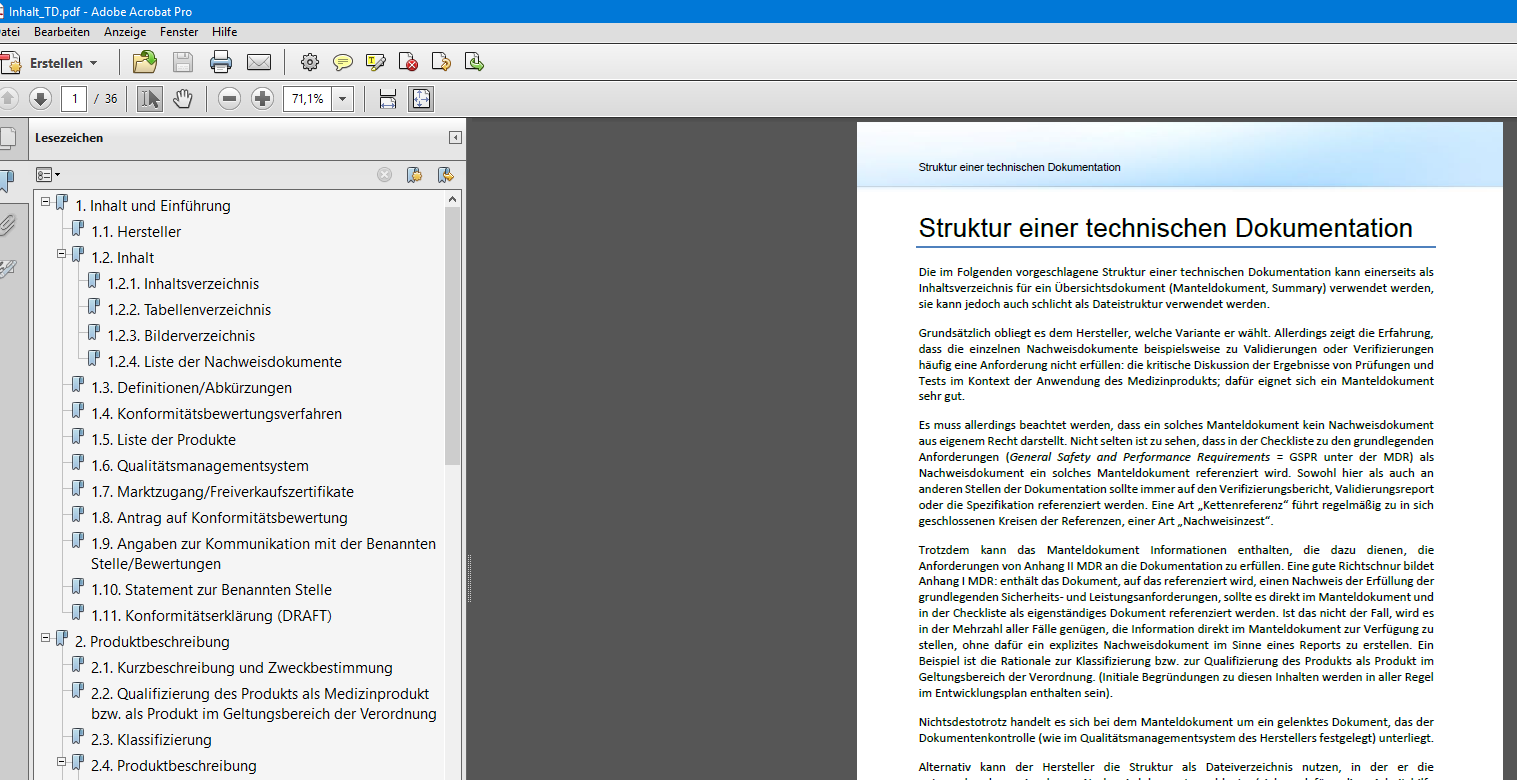

1 Struktur
Anhang IX MDR folgt in seiner Struktur den verschiedenen Aktivitäten bei der Konformitätsbewertung einer Benannten Stelle. Zur Übersicht sei das hier verkürzt dargestellt:
Kapitel I QUALITÄTSMANAGEMENTSYSTEM
In diesem Kapitel werden die Anforderungen an die Antragstellung, das Qualitätsmanagementsystem (nachfolgend kurz: QMS) und seine Dokumentation sowie die Rahmenbedingungen und besonderen Anforderungen an das Audit des QMS sowohl für die initiale Bewertung als auch für die Überwachung während der Laufzeit eines Zertifikats beschrieben.
| • | Abschnitt 1: Grundlagen für die Bewertung des QMS | ||||||||
| • | Abschnitt 2: Bewertung des QMSIn Abschnitt 2 ist die Bewertung des QMS des Herstellers beschrieben. Der Abschnitt ist in vier Unterabschnitte unterteilt, die die unterschiedlichen Aktivitäten für eine Zertifizierung beschreiben:
| ||||||||
| • | Abschnitt 3: Überwachungsbewertung |
Kapitel II BEWERTUNG DER TECHNISCHEN DOKUMENTATION
| • | Abschnitt 4: Bewertung der technischen Dokumentation bei Produkten der Klasse III und der Klasse IIb gemäß Artikel 52 Absatz 4 Unterabsatz 2 MDRAbschnitt 4 beschreibt allgemeingültige Bedingungen für die Bewertung der technischen Dokumentation. | ||||||||
| • | Abschnitt 5: Besondere zusätzliche Verfahren
| ||||||||
| • | Abschnitt 6: Chargenuntersuchung bei Produkten, zu deren integralen Bestandteilen ein Stoff gehört, der für sich allein genommen, als ein aus menschlichem Blut oder Plasma gewonnenes Arzneimittel im Sinne von Artikel 1 Absatz 8 gelten würde.Die Abschnitte 5 und 6 beschreiben die Verfahren für die Bewertung der technischen Dokumentationen für Produkte, wie in Artikel 52 MDR festgelegt. Diese Teile gelten gemeinsam mit den anderen Festlegungen für die zusätzlichen Verfahren, die in Artikel 52 Absätze 9, 10 und 11 MDR sowie in Artikel 54 MDR beschrieben sind. |
Kapitel III VERWALTUNGSVORSCHRIFTEN
| • | Abschnitt 7: Hier werden die Fristen zur Aufbewahrung (und damit zur Bereitstellung der Dokumente für die Behörden) für Hersteller oder Bevollmächtigte geregelt. |
| • | Abschnitt 8: Die Mitgliedstaaten werden in diesem Abschnitt verpflichtet, die Maßnahmen zur Sicherstellung der Anforderungen gemäß Abschnitt 8 für den Fall zu treffen, dass ein Hersteller oder sein Bevollmächtigter vor Ablauf der Fristen in Konkurs geht oder seine Geschäftsfähigkeit aufgibt. |
2 Abschnitt 1.: Grundlagen für die Bewertung des QMS
”Kapitel I: Qualitätsmanagementsystem
1. Der Hersteller richtet ein Qualitätsmanagementsystem gemäß Artikel 10 Absatz 9 ein, das er dokumentiert und umsetzt und für dessen Wirksamkeit während des gesamten Lebenszyklus der betroffenen Produkte er Sorge trägt. Der Hersteller gewährleistet die Anwendung des Qualitätsmanagementsystems nach Maßgabe des Abschnitts 2; er unterliegt Audits gemäß den Abschnitten 2.3 und 2.4 sowie der Überwachung gemäß Abschnitt 3.”
Der erste Abschnitt des Anhangs wiederholt hier die Anforderung aus Artikel 10 MDR, in dem die allgemeinen Anforderungen an einen Hersteller von Medizinprodukten im Sinne eines Wirtschaftsakteurs, beschrieben sind. Die Einzelheiten und Erläuterungen zu den einzelnen Bestandteilen, die in Artikel 10 Abs. 9 MDR aufgeführt sind, finden sich in der Kommentierung zu dem Artikel.
2.1 Qualitätsmanagementsystem EN ISO 13485:2016 vs. MDR
Grundvoraussetzung für die Anwendung des Konformitätsbewertungsverfahrens nach Anhang IX ist also ein vollständiges Qualitätsmanagementsystem (QMS). In aller Regel handelt es sich dabei um ein zertifiziertes QMS nach EN ISO 13485 (Medizinprodukte − Qualitätsmanagementsysteme − Anforderungen für regulatorische Zwecke).
Wichtig
Es ist wichtig zu verstehen, dass ein QMS nach EN ISO 13485 nicht gleichbedeutend mit einem nach MDR zertifizierten QMS ist. Im technischen Report CEN TR 17223 von März 2018 werden beide Regelwerke verglichen, und somit die zusätzlichen Anforderungen der MDR, die von der EN ISO 13485:2016 nicht oder nicht vollständig abgedeckt sind, identifiziert. Im Folgenden findet sich eine kurze Übersicht über die von der EN ISO 13485:2016 nicht oder nicht vollständig umfassten Forderungen der MDR an das Qualitätsmanagementsystem des Herstellers, ohne Anspruch auf detaillierte Vollständigkeit.
Es ist wichtig zu verstehen, dass ein QMS nach EN ISO 13485 nicht gleichbedeutend mit einem nach MDR zertifizierten QMS ist. Im technischen Report CEN TR 17223 von März 2018 werden beide Regelwerke verglichen, und somit die zusätzlichen Anforderungen der MDR, die von der EN ISO 13485:2016 nicht oder nicht vollständig abgedeckt sind, identifiziert. Im Folgenden findet sich eine kurze Übersicht über die von der EN ISO 13485:2016 nicht oder nicht vollständig umfassten Forderungen der MDR an das Qualitätsmanagementsystem des Herstellers, ohne Anspruch auf detaillierte Vollständigkeit.
2.1.1 Risikomanagement
Die MDR stellt an verschiedenen Stellen, besonders jedoch in Anhang I (Grundlegende Sicherheits- und Leistungsanforderungen), konkrete Anforderungen an das Risikomanagement (s. Kap. 02201). Im Vergleich zur EN ISO 13485:2016, die lediglich die Anwendung eines entsprechenden Risikomanagements fordert und dabei auf die EN ISO 14971 referenziert, formuliert die MDR detailliertere Anforderungen u. a.:
| • | Risikomanagement als fortgesetzter iterativer Prozess, der sich über den kompletten Lebenszyklus des Produkts erstreckt und regelmäßige und systematische Aktualisierung erfordert |
| • | Planung, Gefährdungsanalyse, Risikoeinschätzung und Risikobewertung sowohl für den bestimmungsgemäßen Gebrauch als auch für den vorhersehbaren Missbrauch, Beseitigung oder Kontrolle von Risiken, Bewertung von Informationen aus der der Produktion nachgelagerten Phase und ggf. Erweiterung der Risikokontrollmaßnahmen, wenn notwendig. |
| • | Sicherheit durch Herstellung und Auslegung, Schutzmaßnahmen und/oder Alarme sowie Sicherheitsinformationen und ggf. Training der Anwender, um die identifizierten Risiken auf ein akzeptables Minimum zu reduzieren. |
| • | Information der Anwender über alle Restrisiken |
Der im Qualitätsmanagement des Herstellers verankerte Prozess muss die Konformität mit diesen Anforderungen gewährleisten.
2.1.2 Sicherheit und Leistungsfähigkeit im Lebenszyklus
EN ISO 13485:2016 formuliert zwar Anforderungen hinsichtlich der der Produktion nachgelagerten Phasen sowie Produktion, Verpackung und Transport, jedoch wird auch hier die MDR deutlich expliziter, indem sie herausstellt, dass die Produkte auch dann die beschriebenen Charakteristika beibehalten müssen, wenn sie den vorgesehenen Umwelteinflüssen und Belastungen ausgesetzt sind, die für sie beschrieben wurden.
Hersteller sind hier also gefordert, im Rahmen der Verifizierung und Validierung der Auslegung bzw. des Produkts, sicherzustellen, dass das Produkt auch tatsächlich seine beschriebene Leistung erbringt und sicher ist, wenn es wie in den Gebrauchsinformationen beschrieben beispielsweise gelagert oder transportiert wird. Die Erfüllung dieser Anforderung muss durch einen Prozess im Qualitätsmanagementsystem des Herstellers sichergestellt werden. (Beispielsweise bei der Gestaltung des Entwicklungsprozesses).
2.1.3 Klinische Bewertung
Die EN ISO 13485:2016 formuliert auch dafür die Notwendigkeit einer klinischen Bewertung in Übereinstimmung mit den anwendbaren regulatorischen Anforderungen. Eines der großen Kernthemen der MDR ist natürlich die klinische Bewertung, die hier detailliert und maßgeblich die Anforderungen an den Inhalt, die Struktur sowie auch die Häufigkeit der Aktualisierung festlegt.
Auch an dieser Stelle ist das QMS des Herstellers gefordert, einen Prozess zu umfassen, der sicherstellt, dass klinische Bewertungen den Anforderungen der MDR entsprechend durchgeführt werden.
Details dazu finden sich in der Kommentierung zum Artikel 61 MDR (s. Kap. 02061) und Anhang XIV Teil A.
2.1.4 Technische Dokumentation
Die EN ISO 13485:2016 beschreibt im Detail nur die Medizinprodukteakte und die Entwicklungsdokumentation der Medizinprodukte und verweist darüber hinaus lediglich auf eine Dokumentation, um die Erfüllung der regulatorischen Anforderungen nachzuweisen. Die Anforderungen der MDR an diese technische Dokumentation wird in den Anhängen II und III der MDR umfänglich beschrieben (s. Kap. 02202).
Bislang war die Erstellung der technischen Dokumentation häufig eher „historisch gewachsen” und als Prozess bei Herstellern oft nicht beschrieben. Durch die Anforderung in der MDR wird es nun notwendig auch diesen Prozess als geplante und systematische Aktivität im Rahmen des Qualitätsmanagementsystems des Herstellers zu implementieren und zu dokumentieren. Das macht Sinn, denn die Erstellung und die Aktualisierung der technischen Dokumentation gehört zu den Kernelementen der Dokumentation der Konformitätsbewertung.
