
04012 § 12 MPG: Sonderanfertigungen, Medizinprodukte aus Eigenherstellung, Medizinprodukte zur klinischen Prüfung oder für Leistungsbewertungszwecken, Ausstellen
|
Das MPG wurde mit dem Geltungsbeginn der MDR aufgehoben. Allerdings hat es noch so lange Bedeutung, wie die sogenannten Legacy Devices verkehrsfähig sind. Denn die Legacy Devices entsprechen noch den alten Richtlinien 90/385/EWG oder 93/42/EWG und das MPG beinhaltet die nationalen Regeln für die Umsetzung der Richtlinien (s. Kap. 02120, 2.13). Deshalb stellen wir Ihnen die Kommentierung des MPG auch weiterhin in diesem Werk zur Verfügung.
Arbeitshilfen: |

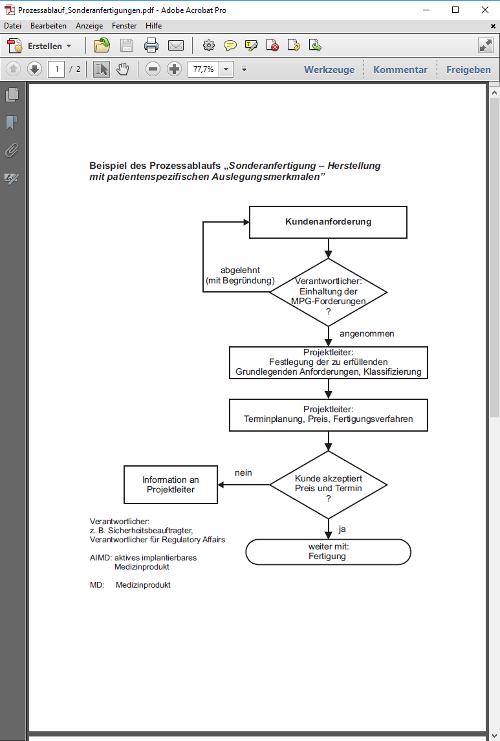

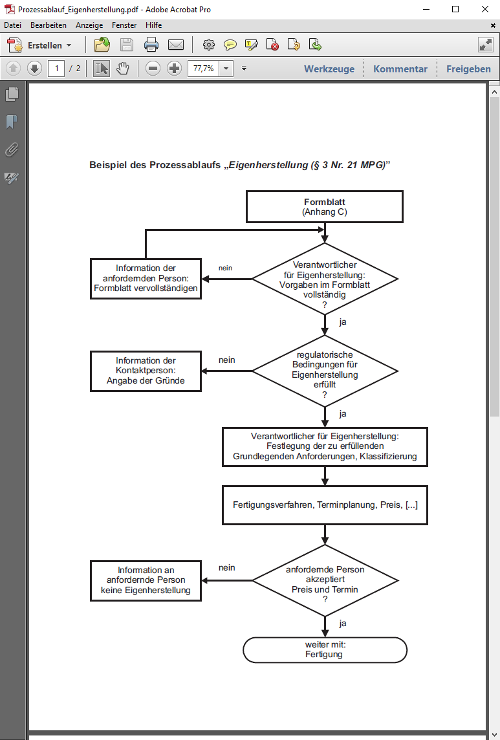
§ 12 Sonderanfertigungen, Medizinprodukte aus Eigenherstellung, Medizinprodukte zur klinischen Prüfung oder für Leistungsbewertungszwecke, Ausstellen
(1) Sonderanfertigungen dürfen nur in den Verkehr gebracht oder in Betrieb genommen werden, wenn die Grundlegenden Anforderungen nach § 7, die auf sie unter Berücksichtigung ihrer Zweckbestimmung anwendbar sind, erfüllt sind und das für sie vorgesehene Konformitätsbewertungsverfahren nach Maßgabe der Rechtsverordnung nach § 37 Abs. 1 durchgeführt worden ist. Der Verantwortliche nach § 5 ist verpflichtet, der zuständigen Behörde auf Anforderung eine Liste der Sonderanfertigungen vorzulegen. Für die Inbetriebnahme von Medizinprodukten aus Eigenherstellung nach § 3 Nr. 21 und 22 finden die Vorschriften des Satzes 1 entsprechende Anwendung.
(2) Medizinprodukte, die zur klinischen Prüfung bestimmt sind, dürfen zu diesem Zwecke an Ärzte, Zahnärzte oder sonstige Personen, die auf Grund ihrer beruflichen Qualifikation zur Durchführung dieser Prüfungen befugt sind, nur abgegeben werden, wenn bei aktiven implantierbaren Medizinprodukten die Anforderungen der Nummer 3.2 Satz 1 und 2 des Anhangs 6 der Richtlinie 90/385/EWG und bei sonstigen Medizinprodukten die Anforderungen der Nummer 3.2 des Anhangs VIII der Richtlinie 93/42/EWG erfüllt sind. Der Sponsor der klinischen Prüfung muss die Dokumentation nach Nummer 3.2 des Anhangs 6 der Richtlinie 90/385/EWG mindestens 15 Jahre und die Dokumentation nach Nummer 3.2 des Anhangs VIII der Richtlinie 93/42/EWG mindestens fünf und im Falle von implantierbaren Medizinprodukten mindestens 15 Jahre nach Beendigung der Prüfung aufbewahren.
(3) In-vitro-Diagnostika für Leistungsbewertungsprüfungen dürfen zu diesem Zwecke an Ärzte, Zahnärzte oder sonstige Personen, die auf Grund ihrer beruflichen Qualifikation zur Durchführung dieser Prüfungen befugt sind, nur abgegeben werden, wenn die Anforderungen der Nummer 3 des Anhangs VIII der Richtlinie 98/79/EG erfüllt sind. Der Sponsor der Leistungsbewertungsprüfung muss die Dokumentation nach Nummer 3 des Anhangs VIII der Richtlinie 98/79/EG mindestens fünf Jahre nach Beendigung der Prüfung aufbewahren.
(4) Medizinprodukte, die nicht den Voraussetzungen nach § 6 Abs. 1 und 2 oder § 10 entsprechen, dürfen nur ausgestellt werden, wenn ein sichtbares Schild deutlich darauf hinweist, dass sie nicht den Anforderungen entsprechen und erst erworben werden können, wenn die Übereinstimmung hergestellt ist. Bei Vorführungen sind die erforderlichen Vorkehrungen zum Schutz von Personen zu treffen. Nach Satz 1 ausgestellte In-vitro-Diagnostika dürfen an Proben, die von einem Besucher der Ausstellung stammen, nicht angewendet werden. |
1 Einleitung
Grundgedanke des Medizinprodukterechts ist der freie Warenverkehr in allen Ländern des Europäischen Wirtschaftsraums. Dabei dürfen Medizinprodukte nur in Verkehr gebracht und in Betrieb genommen werden, wenn
| • | vom Hersteller der Nachweis der Erfüllung der Grundlegenden Anforderungen unter Berücksichtigung der Zweckbestimmung erbracht ist |
und
| • | die Produkte mit der CE-Kennzeichnung versehen sind. |
Für gewisse Produkte – vgl. Anhang 6 der Richtlinie 90/385/EWG „Erklärung zu Geräten für besondere Zwecke”, Anhang VIII der Richtlinie 93/42/EWG „Erklärung zu Produkten für besondere Zwecke”, Anhang VIII der Richtlinie 98/79/EG „Erklärung und Verfahren bei Produkten für Leistungsbewertungszwecken” – legt der Gesetzgeber Sonderregelungen hiervon fest. Diese Sonderregelungen beziehen sich auf Medizinprodukte ohne CE-Kennzeichnung. Diese Sonderregelungen legen fest, unter welchen Bedingungen diese Produkte
